Jürgen Coxa and Gary Kruppab
aResearch Group Lead of Computational Systems Biochemistry, Max Planck Institute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried, Germany. [email protected];  0000-0001-8597-205X
0000-0001-8597-205X
bVice-President, Proteomics, Bruker Daltonics
Over the last two decades, significant advances in technology and new methodologies have made proteomics an extremely powerful tool for protein scientists, biologists and clinical researchers.1 As analytical instrumentation continues to evolve, more data is produced with each technical advancement in proteomics research. That, of course, also creates new challenges for bioinformatics software development.
The modern high-throughput mass spectrometry (MS)-based proteomics methods that are required to gain deeper insights into biological processes produce enormous amounts of data. This raw data necessitates powerful, automated computer-based methods that provide reliable identification and quantification of proteins.
Quantitative proteomics software
Freely available for academic and non-academic researchers, many laboratories across the globe benefit from the MaxQuant quantitative proteomics software package’s precise protein and peptide quantification algorithms. The software was developed by the Computational Systems Biochemistry group at the Max Planck Institute of Biochemistry (MPIB) in Martinsried, Munich, Germany. The group also developed Perseus, a software platform that supports researchers in the interpretation of protein quantification and interaction data as well as data on post-translational modifications (PTMs).
MaxQuant possesses a large ecosystem of algorithms for comprehensive data analysis. It incorporates the peptide search engine Andromeda and, coupled with Perseus, was developed to offer a complete solution for downstream bioinformatics analysis.2 MaxQuant performs quantification with labels and via the MaxLFQ algorithm on label-free data, and achieves high peptide mass accuracies thanks to its advanced non-linear recalibration algorithms.
MaxQuant for four-dimensional proteomics
MaxQuant is often used for liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) shotgun proteomics—a method of identifying proteins in complex mixtures to provide a wider dynamic range and coverage of proteins.
Shotgun (or bottom-up) proteomics is the most commonly used MS-based approach to study proteins by digesting them into peptides prior to MS analysis.
Ion mobility can add a further dimension to LC-MS based shotgun proteomics that has the potential to boost proteome coverage, quantification accuracy and dynamic range. However, this additional information requires suitable software that extracts the information contained in the four-dimensional (4D) data space spanned by m/z, retention time, ion mobility and signal intensity.
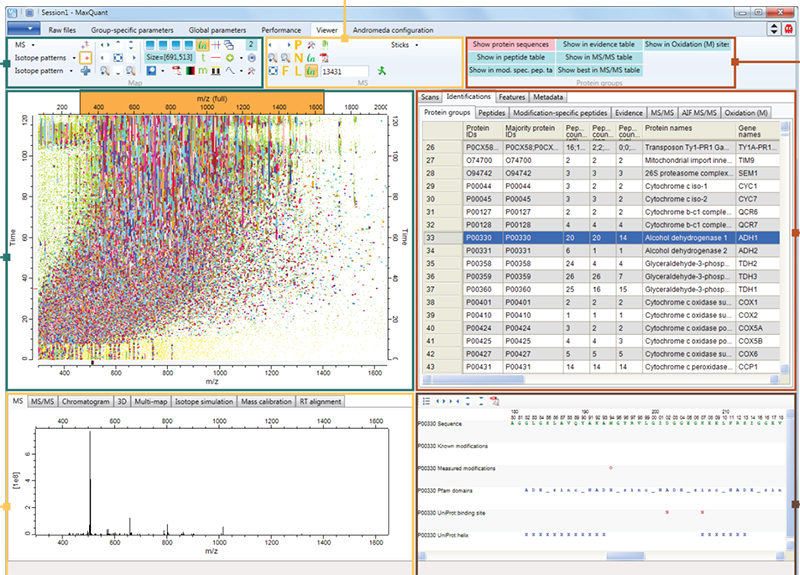
Figure 1. Screenshot of MaxQuant quantitative proteomics software package designed for analysing large mass spectrometric data sets. Source: Maxquant.org
The impact of the addition of the ion mobility dimension on the proteomics field can be seen, for example, in the widespread adoption of Bruker’s timsTOF Pro instrument, which was designed to deliver greater sensitivity, selectivity and MS/MS acquisition speeds for proteomics research. The novel design allows for ions to be accumulated in the front section, while ions in the rear section are sequentially released depending on their ion mobility, and in subsequent scans selected precursors can be targeted for MS/MS. This process is called Parallel Accumulation Serial Fragmentation, or PASEF®.3
The unique trapped ion mobility spectrometry (TIMS) design allows researchers to reproducibly measure the collisional cross-section (CCS) values for all detected ions, and those can be used to further increase the system’s selectivity, enabling more and more reliable relative quantitation information from complex samples and short gradient analyses.
The addition of TIMS to LC-MS based shotgun proteomics, using PASEF, has the potential to boost proteome coverage, quantification accuracy and dynamic range, resulting in fast ultra-sensitive analysis. The increase in speed resulting from PASEF technology allows more samples to be analysed in a shorter time frame, but also generates vast amounts of spectral data, creating challenges when dealing with large sample cohorts.
MPIB software developers adapted the MaxQuant shotgun proteomics workflow to extract this abundance of information from the timsTOF Pro data, making it possible to manage 4D features in the space spanned by retention time, ion mobility, mass and signal intensity that benefit the identification and quantification of peptides, proteins and PTMs.
TIMS is challenging for software developers because it is not just one piece of new information—it adds an additional dimension. The updated MaxQuant 4D-Proteomics workflows can process data produced via PASEF, data-independent acquisition (dia)-PASEF and Mobility Offset Mass Aligned (MOMA).
Adding another dimension can lengthen algorithm processing times, creating a significant challenge for software development with 4D-Proteomics. As a result, the team optimised computation time in MaxQuant to overcome this, so users can achieve good results in a reasonable time frame.
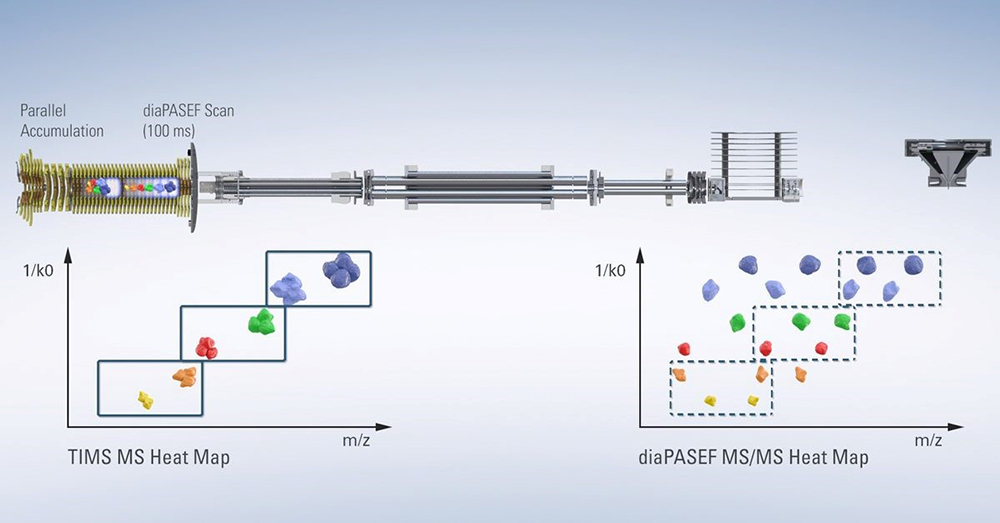
Figure 2. Design of the timsTOF Pro instrument. Source: www.Bruker.com
Emerging applications for 4D-Proteomics
Powerful developments in MS technology have led to the expansion of MaxQuant and the software’s ability to meet future needs in the proteomics field. These improvements are helping researchers develop new capabilities and applications by delivering more sensitivity and selectivity for the identification and quantification of peptides, proteins and PTMs. Instrumentation advances have also bolstered the ongoing development of MaxQuant. The MPIB software development team’s goal is always to expand and improve MaxQuant to meet the complexity of biological processes and novel MS instruments.
Clinical research proteomics
The MPIB Computational Systems Biochemistry team believes clinical research proteomics will be one of the main applications of 4D-Proteomics in the future, and they are working with several clinical groups to bring MS-based proteomics into clinical practice. However, the analysis of proteomics data from samples derived from patients requires special computational strategies. The problems that need to be addressed include: how to extract meaningful protein expression signatures from data with high individual variability, how to integrate the genomic background of the patients into the analysis of proteomics data, and how to determine biomarkers and properly estimate their predictive power.
To answer these questions, the MPIB software developers are working to make use of machine learning algorithms to classify patients and employ feature selection algorithms to extract predictive protein signatures. The extra clinical test engine provides a challenge for the software. It is not clear yet if proteomics will be a guide to which molecules to look at, or if it will be a major component of clinical diagnostics.
Single-cell proteomics
While today’s laboratories generate large data sets from single-cell genomic and single-cell transcriptomic research, single cell proteomics (sc-proteomics) is a nascent field. It holds the potential to enable researchers to detect and quantitate the proteins in single cells, avoiding the need to infer proteins from cellular messenger-RNA levels.4 That also creates new challenges for computational analysis.
MPIB researchers are looking ahead to future needs as sc-proteomics develops, closely examining emerging technologies to establish quantification standards. The software developers believe sc-proteomics will be achievable in the next few years. The two main advances enabling this will be improved sensitivity in the instrumentation and the software to work with it.
Data-independent acquisition
Recent advances in data-independent acquisition (DIA) sensitivity have encouraged the MPIB Computational Systems Biochemistry team to integrate DIA workflows into MaxQuant using machine learning algorithms. The success of DIA relies on key instrumental capabilities—namely resolution, sensitivity, accuracy and dynamic range uncompromised by a fast-spectral acquisition rate.
DIA has been implemented in the timsTOF Pro in a way that takes advantage of the speed and sensitivity of TIMS and PASEF, in a method called dia-PASEF®. The 4D nature of the dia-PASEF data is an advantage for DIA software developers, who can make use of the additional ion mobility dimension for alignment and extraction of features. Such recent technological advances, as well as developments in DIA methods, have provided new opportunities for the MPIB Computational Systems Biochemistry research group.
The MaxQuant developers are enthusiastic about the platform’s upcoming DIA capability. DDA and DIA are becoming comparable because of improved sensitivity in instrumentation. The hardware is becoming simpler for users, but data has been much more challenging because the software must find which fragments belong to which molecule. Addressing these challenges will provide a deeper coverage of proteomics, which could extend the feasibility of applying 4D-Proteomics for clinical research applications, as mentioned previously.
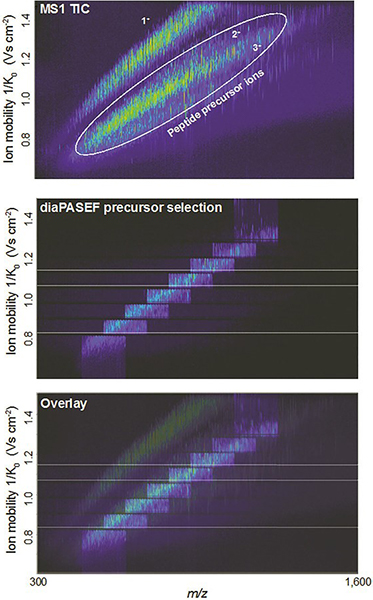
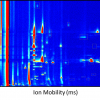
Figure 3. Example showing how dia-PASEF can efficiently fragment nearly all peptide ions eluting at a given retention time. Figures courtesy of: F. Meier, A.-D. Brumer, M. Frank, A. Ha, I. Bludau, E. Voytik, S. Kaspar-Schoenefeld, M. Lubeck, O. Raether, R. Aebersold, B. Collins, H.-L. Rost and M. Mann, “Parallel accumulation – serial fragmentation combined with data-independent acquisition (diaPASEF): Bottom-up proteomics with near optimal usage”, bioRxiv 656207 (2020). https://doi.org/10.1101/656207
Conclusion
Bioinformatics software development is a constantly changing field that will see more technological advancement in the future. As analytical instrumentation advances, the MPIB Computational Systems Biochemistry team must deal with even larger amounts of information on the software side because these instruments continue to expand their dynamic range and capabilities.
These improvements to the MaxQuant software platform are possible due to important collaborations between the MPIB Computational Systems Biochemistry research team and other institutions and industry leaders. These collaborations include projects designed to improve proteomics technologies, with MaxQuant providing the computational tools necessary to analyse data acquired on new and emerging hardware platforms. The results of these collaborations will be used to develop future versions of the software to optimise 4D-Proteomics workflows.
References
- J. Cox and M. Mann, “Quantitative, high-resolution proteomics for data-driven systems biology”, Ann. Rev. Biochem. 80, 273–299 (2011). https://doi.org/10.1146/annurev-biochem-061308-093216
- J. Cox, N. Neuhauser, A. Michalski, R.A. Scheltema, J.V. Olsen and M. Mann, “Andromeda: a peptide search engine integrated into the MaxQuant environment”, J. Proteome Res. 10(4), 1794–1805 (2011). https://doi.org/10.1021/pr101065j
- F. Meier, A.D. Brunner, S. Koch, H. Koch, M. Lubeck, M. Krause, N. Goedecke, J. Decker, T. Kosinski, M.A. Park, N. Bache, O. Hoerning, J. Cox, O. Räther and M. Mann, “Online Parallel Accumulation-Serial Fragmentation (PASEF) with a novel trapped ion mobility mass spectrometer”, Mol. Cell. Proteomics 17(12), 2534–2545 (2018). https://doi.org/10.1074/mcp.TIR118.000900
- V. Marx, “A dream of single-cell proteomics”, Nat. Methods 16, 809–812 (2019). https://doi.org/10.1038/s41592-019-0540-6