
Edwin R. Kellenbach,a Rina K. Dukorb and Laurence A. Nafieb,c
aORGANON BioSciences, PO Box 20, 5340BH Oss, The Netherlands. E-mail: [email protected]
bBioTools Inc., 17546 Bee Line Hwy, Jupiter, FL, 33458, USA
cDepartment of Chemistry, Syracuse University, Syracuse, NY 13244–4100, USA
Introduction
Knowledge of the absolute configuration of asymmetric, chiral carbon atoms is essential for the understanding of enzyme mechanisms, drug action and structure–function relations, as well as for the determination of biological structure.1–3 For example, the right-handed alpha helix in proteins and DNA (both right-handed B and left-handed Z) are all based on the knowledge of the absolute configuration of their building blocks, amino acids and nucleic acids, respectively. Knowledge of the absolute configuration is crucial in the discovery, development and the registration of drugs. The independent determination of the absolute configuration is commonly achieved by single crystal X-ray analysis making use of anomalous dispersion. However, crystals of suitable quality are required for this technique. Moreover, the crystal should preferably contain a heavy atom, such as chlorine or heavier, for the anomalous dispersion technique to work. In this article we describe a technique for determining absolute configurations in solution.
VCD: what is it and how is it used?
Circular dichroism (CD) is the difference in absorption of left and right circularly polarised light.4 Only chiral molecules display CD, and enantiomers have CD of equal magnitude, but opposite sign. In contrast to visible–UV CD, which arises from electronic transitions, VCD originates in vibrational transitions.5–10 As a consequence, even molecules lacking a UV chromophore can be studied, and VCD spectra are much richer in spectral features than electronic CD spectra, analogous to the comparison between UV and IR spectra. Recently, commercial VCD spectrometers have become available. At the same time, developments in quantum chemistry software have allowed the reliable calculation of VCD spectra using density functional theory (DFT).11 Comparison of the observed and calculated VCD spectra provides determination of the absolute configuration of chiral molecules without the need to obtain crystals of the compounds suited for single crystal X-ray analysis. These two developments, VCD instrumentation and software, will increase the application of VCD to stereochemical problems in general and the determination of absolute configuration in particular. In addition, the determination of the absolute configuration by VCD also yields a close approximation of the conformation, or conformational distribution, of the sample molecule in solution.
Equipment
In Figure 1, a schematic overview of a VCD spectrometer is given. Infrared light is passed through a linear polariser to generate linearly polarised light. This is subsequently converted to left and right circularly polarised light by a photo-elastic modulator (PEM).
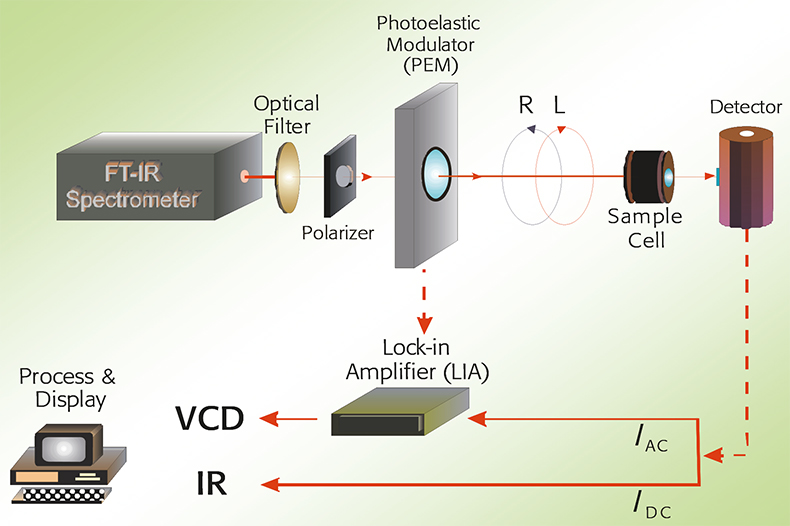
Figure 1. A schematic overview of a VCD spectrometer.
The light is then passed through the sample (either a liquid or a solution) and then to the detector. The VCD signal is demodulated with a lock-in amplifier tuned to the PEM modulation frequency. Both IR and VCD spectra are obtained simultaneously.
Spectra
Figure 2 shows the VCD spectra and IR spectra of (1S) (–)-camphor and (1R)-(+)-camphor.
A number of features immediately stand out:
- VCD spectra display both positive and negative peaks;
- enantiomers have VCD spectra of opposite sign but equal intensity;
- the difference in absorbance between left and right circularly polarised light (ΔA) is very small (10–3–10–5).
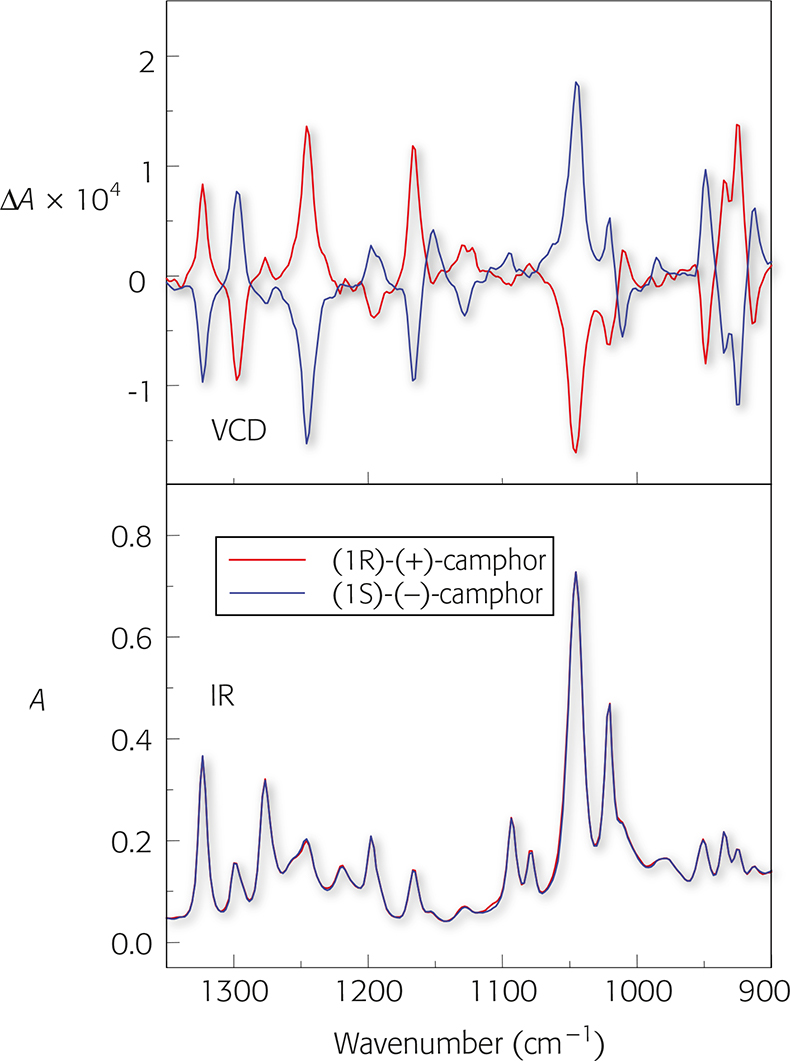
Figure 2. VCD and IR spectra of (1S) (–)-camphor and (1R)-(+)-camphor.
Only (solutions of) chiral molecules give a VCD spectrum. Racemates and non-chiral molecules do not give a signal in VCD. Spectra are often obtained in solutions of carbon tetrachloride (CCl4), deuterated chloroform (CDCl3), dimethyl sulfoxide (DMSO-d6), water (D2O) or as a pure liquid. The solvent is often deuterated to shift the vibrational absorption bands of the solvent away from those of the compound studied. Approximately 5–10 mg of material is required. The measurement time is typically one to several hours, depending on the concentration and the enantiomeric excess. The enantiomeric excess does not have to be 100%, but obviously the VCD bands become weaker and ultimately disappear as the enantiomeric excess approaches zero.
Absolute configuration determination: an example
To determine the absolute configuration of a molecule, the following steps are undertaken:
- The experimental VCD spectrum is measured.
- The VCD of one of the enantiomers is simulated using ab initio DFT methods. The spectrum of the other enantiomer is obtained by reversing the signs of all the VCD bands, or calculating the VCD of the mirror-image structure (opposite enantiomer).
- The experimental spectrum is then compared with the two simulated spectra to see which enantiomer gives the best correlation between signs and intensities.
VCD has been used to determine the absolute configuration of (–)-mirtazapine, the active ingredient of the antidepressant Remeron®,12 which is a racemate where both enantiomers are physiologically active. To obtain the experimental VCD and IR spectra, the (–)-enantiomer was dissolved in CDCl3 at a concentration of 0.10 M (4.5 mg/170 µL solvent). The IR and VCD measurements were carried out with a ChiralIRTM VCD analyser (BioTools, Inc., Jupiter, FL, USA) at a resolution of 8 cm–1, using an IR cell equipped with BaF2 windows and having a pathlength of 72 µm. The VCD spectra were collected as three blocks of 60 minutes each. The blocks were averaged to obtain the final VCD spectrum and appropriately subtracted to obtain the estimate of the noise included in the experimental spectrum. The VCD baseline was obtained by a small correction from the VCD spectrum of the CDCl3 solvent.
To obtain the calculated VCD and IR spectra, the R-enantiomer was first built using HyperChem (Hypercube, Inc., Gainesville, FL, USA) and the geometry optimisation and calculation of vibrational frequencies and IR and VCD intensities were carried out with Gaussian 98 (Gaussian, Inc., Wallingford, CT, USA; Gaussian 03 is now available) at the DFT level (B3LYP functional, 6–31G* basis set). The optimised geometry is shown in Figure 3. The calculations reported here, for a molecule with 39 atoms and 338 basis functions (636 primitive Gaussians) were carried out on a Pentium IV PC, requiring ~30 h cpu. Each step of the geometry optimisation required ~1 h cpu and approximately 1 week of total computer time was required for both geometry optimisation and VCD calculation. The calculated frequencies were scaled by 0.97 and the calculated IR and VCD intensities were converted to Lorenztian bands with 5 cm–1 half-width for visual comparison to the experimental spectra. Both the observed and calculated spectra are shown in Figure 4.
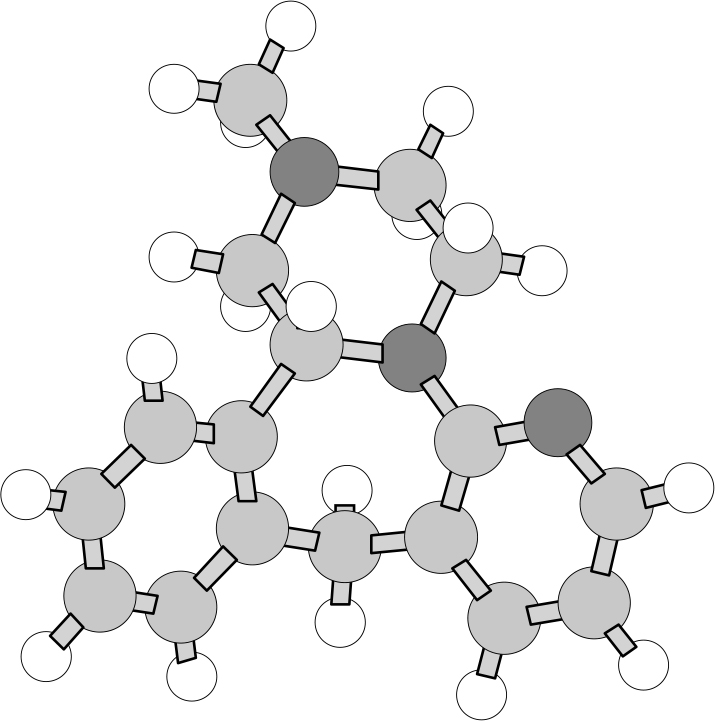
Figure 3. Calculated optimised geometry of R-enantiomer of mirtazapine.
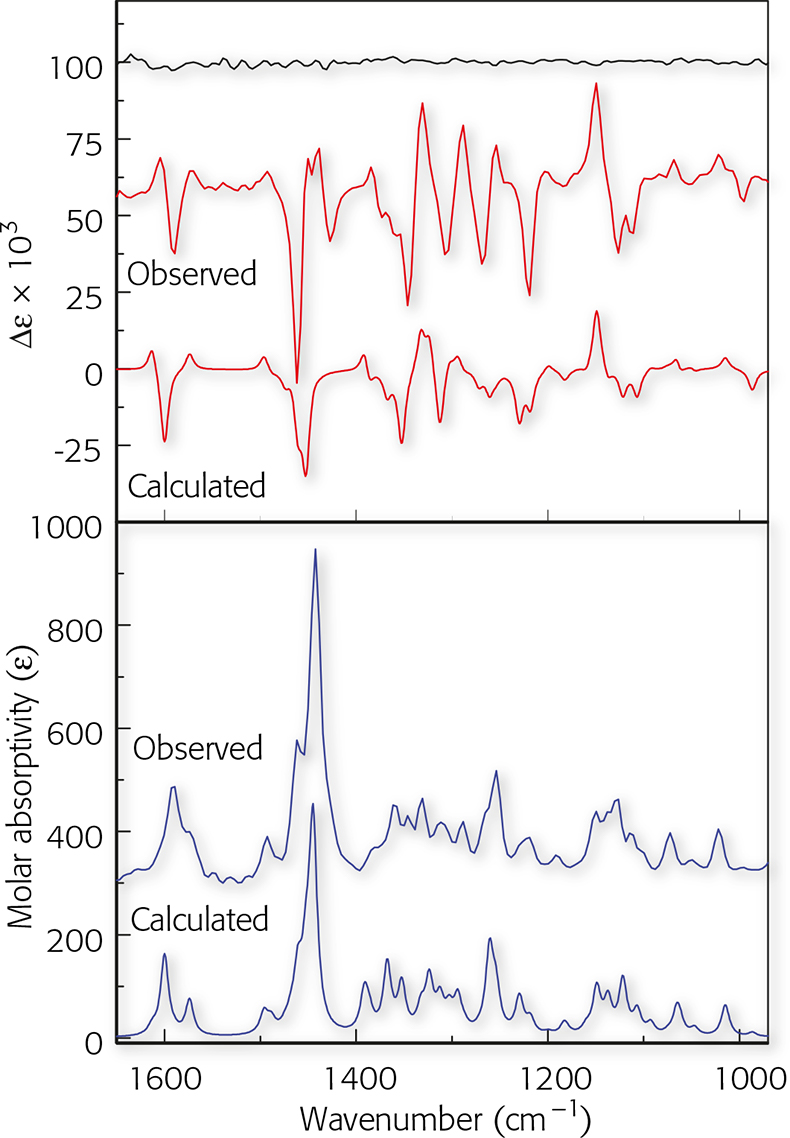
Figure 4. Observed IR and VCD spectra the sample of (−)-mirtazapine compared to calculated spectra for the R-enantiomer. The uppermost trace is the VCD noise level.
The comparison is based on the overall pattern of the relative magnitudes and signs of the corresponding VCD bands in the measured and calculated spectra. In the case of absolute configuration, once a good correlation is established, one only needs to decide whether the signs are all the same or opposite. The knowledge of the absolute configuration used for the calculated VCD spectrum then yields the desired assignment. In this case, the calculated VCD data are in excellent agreement with experiment. For the S-enantiomer, all the VCD bands have opposite sign (but the same magnitude) compared to those calculated for the R-enantiomer. The agreement in sign and relative intensities between the observed and calculated VCD spectra in Figure 4 thus unambiguously assigns the configuration of (–)-mirtazapine as R, and simultaneously identifies the solution conformation in CDCl3. The absolute configuration reported here is in agreement with the one determined independently by synthesis starting from D-(–)-phenylglycine.
The quality of the correspondence is partly due to the rigid structure of mirtazapine, which results in the presence of one single conformer in solution. This substantially simplifies the calculations of the VCD spectrum. For flexible molecules, the calculations are more complex because the VCD of all conformers significantly contributing to the Boltzmann equilibrium distribution need to be calculated and added proportionally. This involves calculating the energies of these conformers relative to one another and sometimes necessitates the inclusion of solvent molecules and/or inter- and intramolecular interactions of the molecule studies. On the other hand, this gives us a unique opportunity to experimentally verify the calculated conformers and their populations by the comparison of observed and calculated VCD. To make the comparison more objective a calculated similarity measure can be used.13 In contrast to nuclear magnetic resonance (NMR), which yields weighted averages for fast molecular movements, VCD detects each species present as a linear superposition of all contributing conformers.
Conclusion
VCD is an excellent technique for the determination of the absolute configuration and conformation of small and medium sized molecules in solution. Increase in computer calculation speed will result in an extension of this technique to larger and more flexible molecules and will allow more efficient calculation of VCD spectra.
References
- C. McManus, Right Hand, Left Hand. Phoenix Publishers, Phoenix (2003).
- M. Gardner, The New Ambidextrous Universe: Symmetry and Asymmetry for Mirror Reflections to Superstrings. Dover Publications, Mineola (2005).
- J. Jacques, La Molecule et son Double. Hachette Book Group, New York (1992).
- P. Crabbé, Optical Rotary Dispersion and Circular Dichroism in Organic Chemistry. Holden-Day, San Francisco (1965).
- L.A. Nafie, “Infrared and Raman Vibrational Optical Activity: Theoretical and Experimental Aspects”, Ann. Rev. Phys. Chem. 48, 357–386 (1997). https://doi.org/10.1146/annurev.physchem.48.1.357
- L.A. Nafie and T.B. Freedman, “Vibrational Circular Dichroism: An Incisive Tool for Stereochemical Applications”, Enantiomer 3, 283–297 (1998).
- R.K. Dukor and L.A. Nafie, “Vibrational Optical Activity of Pharmaceutical Molecules”, in Encyclopedia of Analytical Chemistry, Ed by R.A. Meyers. John Wiley & Sons, Chichester, pp. 662–676 (2000). https://doi.org/10.1002/9780470027318.a0215
- P.J. Stephens and F.J. Devlin, “Determination of the structure of chiral molecules using ab initio vibrational circular dichroism spectroscopy”, Chirality 12(4), 172–179 (2000). https://doi.org/10.1002/(SICI)1520-636X(2000)12:4<172::AID-CHIR3>3.0.CO;2-6
- T.B. Freedman, X. Cao, R.K. Dukor and L.A. Nafie, “Absolute configuration determination of chiral molecules in the solution state using vibrational circular dichroism”, Chirality 15(9), 743–758 (2003). https://doi.org/10.1002/chir.10287
- T. Kuppens, P. Bultinck and W. Langenaeker, “Determination of absolute configuration via vibrational circular dichroism”, Drug Discovery Today: Technologies 1(3), 269–275 (2004). https://doi.org/10.1016/j.ddtec.2004.11.004
- J. R. Cheeseman, M.J. Frisch, F.J. Devlin and P.J. Stephens, “Ab initio calculations of atomic axial tensors and vibrational rotational strengths using density functional theory”, Chem. Phys. Lett. 252(3,4), 211–220 (1996).
- T.B. Freedman, R.K. Dukor, P.J.C. van Hoof, E.R. Kellenbach and L.A. Nafie, “Determination of the absolute configuration of (–)-mirtazapine by vibrational circular dichroism”, Helvetica Chim. Acta 85(4), 1160–1165 (2002). https://doi.org/10.1002/1522-2675(200204)85:4<1160::AID-HLCA1160>3.0.CO;2-T
- T. Kuppens, K. Vandyck, J. Van der Eycken, W. Herrebout, B.J. van der Veken and P. Bultinck, “Determination of the Absolute Configuration of Three as-Hydrindacene Compounds by Vibrational Circular Dichroism”, J. Org. Chem. 70(23), 9103–9114 (2005). https://doi.org/10.1021/jo050666h


