Eva Marguí,a* Clàudia Fontàs,a Katleen Van Meel,b Manuela Hidalgoa and Ignasi Queraltc
aDepartment of Chemistry, University of Girona, Campus Montilivi, 17071 Girona, Spain
bDepartment of Chemistry, University of Antwerp, Universiteitsplein 1, 2610 Antwerp, Belgium
cLaboratory of X-Ray Analytical Applications, Institute of Earth Sciences “Jaume Almera”, CSIC, Solé Sabarís s/n, 08028 Barcelona, Spain
One of the dangerous kinds of pollution in aquatic systems is due to the dumping of materials containing heavy metals. Hence, the monitoring of heavy metals in aqueous samples is becoming increasingly important. A promising method to analyse such samples is a combination of preconcentration and X-ray fluorescence analysis.
Introduction
One of the dangerous kinds of pollution in aquatic systems is due to the dumping of materials containing heavy metals. Hence, the monitoring of heavy metals in aqueous samples is becoming increasingly important.
Normally, metal concentrations in water are in the ng L–1 range, and the analytical procedures used for their determination are usually based on Anodic Stripping Voltametry (ASV) and Atomic Spectrometry, including Electrothermal Atomic Absorption Spectrometry (ETAAS), Inductively Coupled Plasma Atomic Emission Spectrometry (ICP-AES) and Inductively Coupled Plasma Mass Spectrometry (ICP-MS).
However, the direct analysis of some complex environmental samples like seawater presents some difficulties, mainly due to the high salinity of the matrix. Therefore, in such cases, a dilution of the sample may be necessary before the analysis, or a preliminary separation and/or preconcentration step may be required to eliminate interferences and/or to improve detection limits for metals in the low µg L–1 range. Moreover, when the analysis is performed by using solid sorbents followed by spectrophotometric techniques, an additional elution step after the preconcentration procedure is necessary to recover the species in an appropriate medium.
A promising alternative is a combination of preconcentration and X-ray fluorescence (XRF) analysis. XRF spectrometry is a popular method for the direct determination of major and minor elements in mineralogical and environmental solid samples. Using this technique, the direct quantitation of metal species held in solid materials is possible and therefore the elution step can be avoided, leading to a reduction of sample handling.
Several sorbents have been reported in the literature to preconcentrate metals prior to their determination by XRF techniques,1 such as ion-exchange materials and polyurethane foam. Another possibility widely used for preconcentration purposes is the formation of insoluble precipitates of the metal of interest by adding pyrrolidinedithiocarbamates and the collection of the precipitate on cellulose filters. Nevertheless, these XRF methods involve the addition of reagents, adjusting the pH of test samples, laborious filtrations and drying stages, with the possibility of contamination of samples during the sample preparation. Furthermore, sometimes it is difficult to obtain homogeneous specimens with satisfactory working surfaces after water evaporation or concentration of trace elements by precipitation or coprecipitation methods.
In this article, a method is outlined for the determination of trace amounts of metal contaminants in saline and acidic solutions based on a preconcentration with membranes followed by XRF detection. The method is based on the principle of metal preconcentration using membranes containing the commercial anion-exchanger tricaprylylmethylammonium chloride (Aliquat 336). This is a particularly convenient means of separation for the XRF method because the membrane can be mounted directly in the spectrometer for analysis, thereby offering a novel and easy alternative to the complicated preconcentration methods described above when dealing with this type of matrix. Moreover, with the preconcentration procedure proposed, the problems of reduced sensitivity inherent to the choice of La lines for the measurement of high atomic number elements (commonly used in conventional XRF instrumentation) are reduced and the determination of some important pollutant elements is possible in the mg L–1 range.
In particular, the feasibility of the combined use of this simple and inexpensive membrane preconcentration procedure with different configurations of XRF spectrometers [energy dispersive XRF (EDXRF), wavelength dispersive XRF (WDXRF), high-energy-polarised-energy dispersive XRF (HE-P-EDXRF)] for the determination of trace amounts of some metallic pollutants (Cd, Cr, Pd and Pt) in complex liquid samlpes is outlined in this article. Detailed information and results about this topic are described in full in previously published papers.2–3
Analysis of liquid samples by XRF
Frequently, direct XRF analysis of solutions entails technical difficulties and is characterised by particular errors in the results. Two general methods of analysis of non-volatile elements in liquid samples are commonly used: (i) as a liquid deposited in a special liquid sample holder, which incorporates a tapered snap-on ring at the end of the cell for attachment of a thin-film liquid sample support or (ii) evaporated onto a support under vacuum. When using the first method, bubbles released from solutions due to inadequate sample holder filling and heating of the solution can give certain problems. Moreover, liquid samples usually provide a high X-ray scatter background resulting in poor signal-to-noise ratio. Typical detection limits of conventional direct XRF are in the mg L–1 range, which is not satisfactory for environmental requirements. The sensitivity can be improved using Total Reflection X-Ray Fluorescence spectrometry (TXRF). The TXRF system makes use of the fact that, at very low glancing angles, the high background that would generally occur due to scatter from the sample support is absent. Because the background is so low, concentrations in the range of several µg L–1 can be measured in aqueous samples such as drinking water, rainwater and stream waters. However, when dealing with the analysis of wastewater and seawater, a special preparation method has also to be applied to separate the suspended matter and to remove the salt content prior to the measurement. Moreover, this kind of X-ray spectrometer may not be generally available to the general user community.
The simple method described here, as mentioned above, is based on the principles of metal preconcentration using membranes containing the commercial anion-exchanger Aliquat 336.
The extraction ability of the Aliquat 336 (R3R’N+Cl–) is based on the formation of an ion-pair in the membrane between the cationic part of the quaternary ammonium salt and the corresponding anionic metal complex. In general, the extraction can be described as follows:
MXnm–(aq) + m R3R’N+Cl–(memb) ® (R3R’N+)m MXn (memb) + mCl–(aq)
where MXnm– represents the anionic metal complex (being in this case: HCrO4–, CdCl42–, PdCl42–, PtCl62–) and aq and memb denote aqueous phase and membrane support, respectively.
For the preparation of the membranes, a polyvinylidene difluoride (PVDF) film (Durapore, Millipore; average pore size 0.2 µm, porosity 75% and average thickness 125 µm) was used to contain the extractant solution. For this, the polymeric support was soaked in a solution of 500 mM of Aliquat 336 in decaline for 10 min and then taken out of the organic solution and wiped with a piece of filter paper. The organic solution remained in the pores of the PVDF support by capillarity.
The membrane preconcentration experiments were carried out by contacting the aqueous solutions of each metal with the impregnated thin layers under continuous stirring. At the end of the experiments, the loaded membranes were removed from the vessel, washed with deionised water several times, and allowed to dry at room temperature before XRF analysis. In Figure 1, a photo of some membranes containing different amounts of metal are shown.

Figure 1. Picture of membranes after being put in contact with liquids containing, from left to right, increasing amounts of Pd.
XRF instrumentation
There are many types of XRF spectrometers available on the market today, most of which can be separated roughly into two categories: wavelength dispersive X-ray fluorescence and energy dispersive X-Ray fluorescence. In the study reported here, three different XRF spectrometers were used for the determination of the metals loaded in the membranes. Full details on instrumental parameters and measurement conditions for each spectrometer are given elsewhere.2–3
Theoretical principles of the XRF technique and detailed instrumental characteristics were described in a previous recently published work in Spectroscopy Europe in which the feasibility of the XRF technique was demonstrated for the determination of chemical composition of vegetation samples.4
Evaluation of membrane preconcentration procedure
In a first stage, the effectiveness and applicability of using the membranes as a preconcentration procedure was evaluated. For that, the extraction efficiency versus time was evaluated for each metal by contacting different membranes with the aqueous solutions at different metal concentrations. The metal extraction was followed by analysis by ICP-AES of samples withdrawn at different times and results obtained in terms of metal extracted. For all studied metals, the extraction efficiency increased with time reaching a constant value of extraction that was different for each metal [Cr(VI): 90 min, Cd(II): 360 min, Pd(II) and Pt(IV): 120 min]. Furthermore, it was observed that the extraction for a given metal was independent of the initial metal concentration leading to the conclusion that the metal collected on the membranes was related to the initial metal content in aqueous solutions. This fact points out the viability of using these membranes as standards for liquid sample preparation in XRF analysis.
In a second set of experiments, the homogeneity of metal loaded in the membranes was also conducted. As an example, Figure 2 shows a mapping of Cd distribution using the Fischer XDAL EDXRF spectrometer.
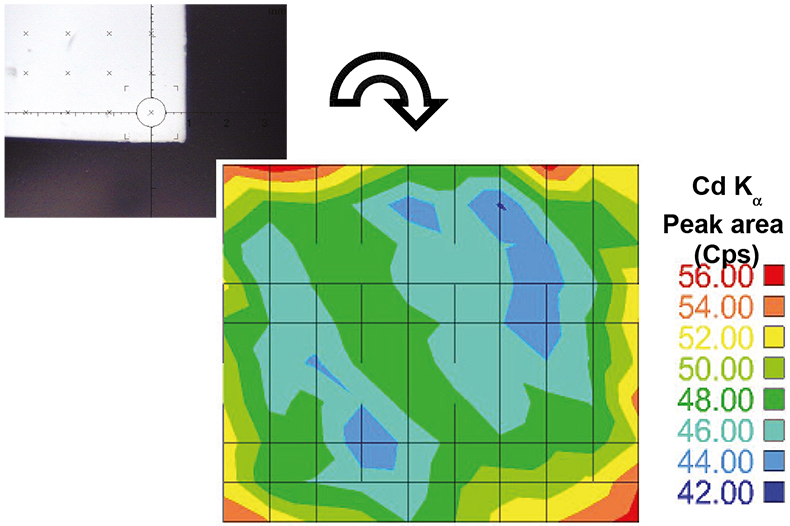
Figure 2. Mapping of Cd distribution on a loaded membrane (Fischer XDAL EDXRF spectrometer) using a measuring routine of 10 × 10 points.
Analytical performance of membrane-XRF system
Spectra of the metal collected in the membranes (previously put in contact with aqueous solutions containing different amounts of metal) were recorded under the selected operating conditions. It was found that the XRF signal increased when increasing the metal concentration initially present in the aqueous solution. As is shown in Figure 3, the representation of the peak area obtained for each activated thin layer vs the initial Cr(VI) content in the aqueous solution (in the range of 0.42–8.82 mg L–1 Cr) shows a very good correlation, indicating that this relationship can be used as a calibration curve for future determination of Cr(VI) when contained in liquid samples. In Table 1, the analytical performance of the membrane-XRF system for each metal and studied working range is summarised. Acceptable correlation coefficients were achieved in all cases indicating a good linearity for the studied working range under the experimental conditions employed. This fact confirms the suitability of using these membranes as a standard sample preparation method for XRF analysis since the metal collected on them (expressed as mg) could be related with the initial metal content in the aqueous solution (mg L–1). Moreover, the use of membranes avoids the need for matrix and thickness effect corrections, commonly required in XRF analysis, since in this case, the attenuation of the incident primary and emergent secondary spectral line radiation is almost negligible and, thus, the XRF signal is proportional to the metal concentration.
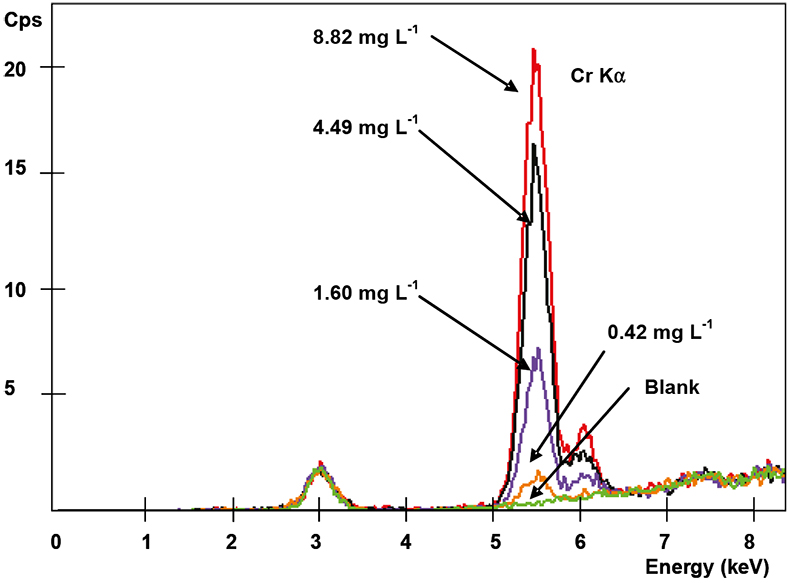
Figure 3. Spectra of Cr(VI) collected on membranes at different initial metal concentration solutions (Fischer XDAL EDXRF spectrometer).
Table 1. Analytical performance for Cd, Cr, Pd and Pt determination using membrane preconcentration plus XRF analysis.
Metal | Instrument | Concentration range (mg L–1) | Regression coefficient | LOD (mg L–1) |
Cr | Fischer XDAL EDXRF | 0.42–8.8 | 0.962 | 0.22 |
Pd | Bruker S4 Explorer WDXRF | 1–50 | 0.994 | 0.015 |
Pt | Bruker S4 Explorer WDXRF | 1–30 | 0.970 | 0.014 |
Cd | Fischer XDAL EDXRF | 0.5–50 | 0.997 | 0.34 |
Epsilon 5 HE-P-EDXRF | 10–100* | 0.993 | 1.20* |
*Concentrations are expressed as µg L–1
Notes: LOD = Limit of detection, calculated as three times the standard deviation of the background
Table 2. Cd concentrations in spiked seawater determined by using an Epsilon 5 HE-P-EDXRF and Anodic Stripping Voltametry (ASV).
Cadmium added (µg L–1) | HE-P-EDXRF | ASV | |
Al2O3 target | CsI target | ||
20 100 600 | 20 (s = 2) 102 (s = 3) 553 (s = 79) | 21 (s = 3) 100 (s = 5) 582 (s = 76) | 26 (s = 3) 106 (s = 4) 617 (s = 6) |
s: standard deviation for duplicate spiked seawater samples
The calculated detection limits, in all cases, were in the µg L–1 range, indicating the possibilities of the outlined methodology for trace determination content of Cd, Cr(VI), Pd and Pt in liquid saline solutions using conventional EDXRF and WDXRF instrumentation. From Table 1 the benefits of using polarised X-rays sources (Epsilon 5 HE-P-EDXRF) can be appreciated, since the detection limits obtained for Cd are about one order of magnitude lower than those attained for conventional XRF instrumentation and, moreover, they were also competitive with those determined using other popular spectrometric techniques such as flame-AAS (FAAS) and ICP-AES. In order to test the suitability of the method when dealing with complex matrices, the determination of metal in spiked seawater samples was undertaken. As Table 2 shows, the results obtained for Cd with the outlined analytical method were similar to those obtained using the ASV technique, which is commonly used for the analysis of metals in high salinity solutions, and no significant differences at the 95% confidence level were found. Thus, the accuracy of the procedure and the absence of matrix effects were confirmed for this type of high salinity samples.
The precision of the methodology was evaluated in terms of relative standard deviation (RSD) of replicate analysis of standard solutions and at different initial metal concentrations. In all cases, relative standard deviations calculated were less than 10%.
A study was conducted to compare the analytical improvements of the proposed methodology with the direct analysis of saline solutions. For this purpose, an aliquot of 7 mL of a spiked seawater sample was deposited in the special liquid sample holder and analysed directly with the HE-P-EDXRF spectrometer using the same instrumental conditions as for the membrane analysis. In Figure 4, the spectra obtained for the direct analysis of a seawater sample containing 600 µg L–1 of Cd and after applying the developed preconcentration step are displayed. As can be seen, a very intense peak is found in the case of the membranes preconcentration, while in the case of the direct analysis of the liquid sample the peak is obviously below the detection limit. From the same figure, the reduction of the background spectrum can be appreciated when using the preconcentration procedure. Therefore, it is clear that the method described offers huge benefits in terms of sensitivity since the LoDs are improved by several orders of magnitude compared with the direct analysis of seawater.
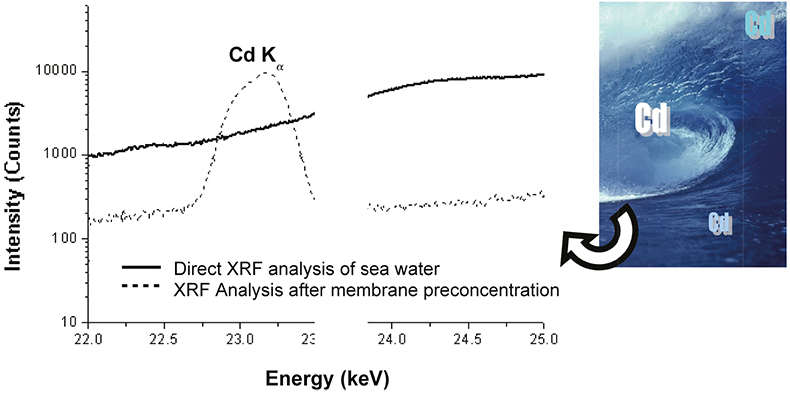
Figure 4. Comparison between spectra obtained for the direct analysis of a seawater sample containing 600 µg L–1 of Cd and after membrane preconcentration step. (Epsilon 5 HE-P-EDXRF, secondary target: CsI).
Summary
The data reported highlights that the use of XRF spectrometry, in particular HE-P-EDXRF, in combination with membrane preconcentration is a promising alternative for metal analysis at low µg L–1 levels in complex environmental liquid samples.
The method described offers important advantages, such as simplicity of operation and high throughput, compared to other methodologies.
Acknowledgements
This study was financed by the Spanish National Research Programme (projects CGL2004–05963-C04/HID and CGL2007–66861-C04). Authors want to thank Dr V. Rossiger from Helmut Fischer GmbH+Co and Mr Joan Pujol (Fischer Instruments, Spain) for their technical support during the present research. Antonio Jurado and Ester Sagristà are acknowledged for their valuable support in the laboratory work.
References
- R. Van Grieken, Anal. Chim. Acta 3 (1982).
- C. Fontàs, I. Queralt and M. Hidalgo, Spectrochim. Acta Part B 407 (2006).
- E. Marguí, C. Fontàs, K. Van Meel, R. Van Grieken, I. Queralt and M. Hidalgo, Anal. Chem., in press (doi: 10.1021/ac7018427).
- E. Marguí, M. Hidalgo and I. Queralt, Spectrosc. Europe 19(3), 13 (2007).